尋媶徯夘
1乯恄宱桼棃嵶朎奜彫朎暘棧媄弍偺奐敪乗Brain Liquid Biopsy媄弍偺奐敪
丂嵶朎奜彫朎乮EV:僄僋僜僜乕儉傪娷傓乯偼條乆側嵶朎偐傜暘斿偝傟丄寣塼側偳偺懱塼拞偵懚嵼偡傞偲偝傟傞丅拞悤恄宱嵶朎傕椺奜偱偼側偔丄恄宱桼棃偺EV(neuron-derived EV:NDE)偑枛徑寣拞偵暘斿偝傟偰偄傞偲峫偊傜傟傞丅
EV偼帀幙擇廳枌偵暍傢傟丄偦偺拞恎丄偡側傢偪抈敀丄DNA丄RNA側偳偼枛徑傑偱曐懚偝傟偰偄傞丅廬偭偰丄摿掕偺嵶朎偺EV傪暘棧偱偒傟偽丄暘斿尦偺嵶朎偺娐嫬傪暘愅偱偒傞丅偮傑傝丄NDE傪枛徑寣偐傜暘棧偱偒傟偽丄拞悤恄宱偺忬嫷傪夝愅偱偒傞偙偲偵側傞丅
偦偙偱丄変乆偼枛徑寣偐傜EV枌昞柺偵偁傞摿庩側恄宱摿堎揑側抈敀乮MPX乯偵懳偡傞峈懱傪梡偄丄柶塽捑崀朄偵傛傞NDE暘棧朄傪奐敪偟偰偄傞乮恾嶲徠乯丅
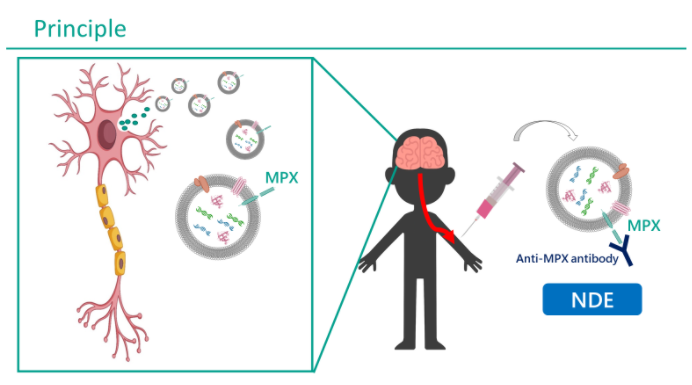 嬤擭丄偑傫恌抐椞堟偱偼枛徑寣偺EV側偳傪梡偄偨Liquid Biopsy偑奐敪偝傟丄偑傫慻怐傪捈愙Biopsy偡傞偙偲側偔丄旕怤廝揑偵偑傫恌抐偑峴偊傞傛偆偵側傝偮偮偁傞丅偦偰偵側偧傜偊丄変乆偼Brain Liquid Biopsy傪採埬偡傞丅偡側傢偪枛徑寣偺NDE傪暘棧偟夝愅偡傞偙偲偱丄惛恄恄宱幘姵偺僶僀僆儅乕僇乕偺妋棫傪栚巜偟偰偄傞丅偝傜偵丄Brain Liquid Biopsy偼惛恄恄宱幘姵偺昦懺夝柧丄偝傜偵偼憂栻僞乕僎僢僩偺扵嶕偑壜擻偲峫偊偰偄傞丅
偙偺尋媶偼丄擔杮堛椕尋媶奐敪婡峔乮AMED乯乽僸僩擼桼棃僄僋僜僜乕儉傪棙梡偟偨擣抦徢姵幰傪憌暿壔偡傞庤朄偺奐敪尋媶乮尋媶奐敪戙昞幰:岺摗嫪乯乿偲偟偰峴傢傟偰偄傞丅
嬤擭丄偑傫恌抐椞堟偱偼枛徑寣偺EV側偳傪梡偄偨Liquid Biopsy偑奐敪偝傟丄偑傫慻怐傪捈愙Biopsy偡傞偙偲側偔丄旕怤廝揑偵偑傫恌抐偑峴偊傞傛偆偵側傝偮偮偁傞丅偦偰偵側偧傜偊丄変乆偼Brain Liquid Biopsy傪採埬偡傞丅偡側傢偪枛徑寣偺NDE傪暘棧偟夝愅偡傞偙偲偱丄惛恄恄宱幘姵偺僶僀僆儅乕僇乕偺妋棫傪栚巜偟偰偄傞丅偝傜偵丄Brain Liquid Biopsy偼惛恄恄宱幘姵偺昦懺夝柧丄偝傜偵偼憂栻僞乕僎僢僩偺扵嶕偑壜擻偲峫偊偰偄傞丅
偙偺尋媶偼丄擔杮堛椕尋媶奐敪婡峔乮AMED乯乽僸僩擼桼棃僄僋僜僜乕儉傪棙梡偟偨擣抦徢姵幰傪憌暿壔偡傞庤朄偺奐敪尋媶乮尋媶奐敪戙昞幰:岺摗嫪乯乿偲偟偰峴傢傟偰偄傞丅
2乯AI傪梡偄偨懳榖偵傛傞憗婜擣抦徢専抦僔僗僥儉偺奐敪
丂崱屻丄擣抦徢偵懳偡傞幘姵廋忺栻偺椪彴摫擖傪峊偊丄擣抦徢偺憗婜恌抐偼媔嬞偺壽戣偱偁傞丅偙偺傛偆側忬嫷偺拞丄堛椕婡娭偱偺専嵏偵偮側偑傞娙曋側擣抦徢専抦僔僗僥儉偺奐敪偑朷傑傟傞丅
変乆偼夋柺忋偺傾僶僞乕偑條乆側幙栤傪旐専幰偵搳偘偐偗丄偦偺摎偊曽偺敪岅忬嫷傗昞忣傪AI偵婡夿妛廗偝偣偰丄擣抦徢傪専抦偡傞僔僗僥儉傪奐敪偟偰偄傞丅傾僶僞乕偼擔忢揑側幙栤傪偡傞偺偱丄旐専幰偼専嵏傪庴偗偰偄傞偲姶妎偱偼側偔丄婥寉偵懳榖傪偡傞偲偄偆偙偲偱丄擣抦徢偺専抦偑壜擻偱偁傞乮摦夋嶲徠http://www.autism-communication.com/~hiroki-tan/avatar_demo.mp4 乯丅偙傟偼擣抦徢偺侾偮偺僶僀僆儅乕僇乕偲偄偊傞丅
杮尋媶偼丄撧椙愭抂壢妛媄弍戝妛堾戝妛偲偺嫟摨尋媶偱偁傞丅
3乯偆偮昦偺暘巕惗暔妛揑昦懺夝柧媦傃帯椕朄奐敪
丂摉尋媶幒偱偼廬棃偐傜彫朎懱僗僩儗僗乮仏乯偲偆偮昦偺昦懺夁掱偲偺娭楢偵偮偄偰専摙偟偰偒偨丅嬤擭丄彫朎懱僗僩儗僗偵傛偭偰僔僌儅1庴梕懱偑桿摫偝傟傞偙偲傪敪尒偟丄偙偺儊僇僯僘儉偲偆偮昦偵偍偗傞敀幙忈奞偺娭楢偵偮偄偰専摙傪恑傔偰偄傞丅幚尡庤朄偲偟偰偼丄嵶朎幚尡傗堚揱巕夵曄儅僂僗傪巊偭偨僗僩儗僗晧壸幚尡側偳偑拞怱偱偁傞丅
仏彫朎懱僗僩儗僗斀墳
侾丏俁偮偺彫朎懱僗僩儗僗斀墳
嵶朎彫婍姱偺侾偮偱偁傞彫朎懱乮ER乯偼丄暘斿抈敀傗枌峔惉抈敀側偳偺愜傝偨偨傒傗東栿屻廋忺傪偍偙側偆抈敀偺乽慻傒棫偰岺応乿偺傛偆側栶妱傪扴偆丅乽慻傒棫偰岺応乿偑屘偵乽晄椙昳乿懄偪愜傝偨偨傒偑晄廫暘側埥偄偼晄惓抈敀(unfolded protein)偺弌尰偼廻柦偺傛偆側傕偺偱偁傞丅僇儖僔僂儉摦懺偺曄壔丄巁壔娨尦忬懺偺曄壔丄暘斿抈敀偺夁忚嶻惗丄僽僪僂摐寚朢丄摐晅壛偺曄壔側偳偺嵶朎撪僗僩儗僗偼ER僗僩儗僗偲尵傢傟丄ER撪偺unfolded protein偺憹壛傪棃偡丅偦偺傛偆側乽晄椙昳乿偑乽弌壸乿偝傟側偄傛偆偵ER偵偼unfolded protein response (UPR)偲尵偆乽昳幙娗棟乿婡擻傪桳偡傞丅尰嵼偱偼俁偮偺ER僗僩儗僗斀墳乮UPR乯偑憐掕偝傟偰偄傞丅嵶朎偼偙傟傜偺婡峔偵傛偭偰ER僗僩儗僗傪崕暈偟傛偆偲偡傞偑丄壗傜偐偺棟桼偱僗僩儗僗偑崕暈偝傟側偄偲嵶朎偼屻弎偡傞傾億僩乕僔僗宱楬傊摫偐傟傞丅
倎丏抈敀東栿梷惂乮Translational attenuation乯
戞侾偺愴棯偲偟偰丄unfolded protein偑ER撪偵偙傟埲忋拁愊偟側偄傛偆偵丄嵶朎偼丄抈敀東栿慡斒傪梷惂偡傞偲偄偆曽嶔傪島偠傞丅偙傟偼丄東栿奐巒場巕eIF2兛偺儕儞巁壔偵傛偭偰傕偨傜偝傟傞乮Shi et al, 1998; Harding et al,1999乯丅
倐丏僔儍儁儘儞抈敀桿摫
戞俀偺愴棯偲偟偰丄嵶朎偼ER僗僩儗僗偵傛傞unfolded protein偺ER撪偱偺拁愊傪嶡抦偟丄ER偐傜妀傊嵶朎撪僔僌僫儖揱払傪妶惈壔偝偣丄僔儍儁儘儞抈敀偱偁傞BiP丄calnexin丄傗calreticulin側偳傪敪尰桿摫偡傞丅偙傟傜僔儍儁儘儞抈敀偼丄ER偵拁愊偟偨unfolded protein偺愜傝偨偨傒傪懀恑偁傞偄偼惀惓偡傞(Sidrauski et al, 1998)丅
們丏彫朎懱娭楢暘夝 (ER-associated degradation :ERAD)
ER偵拁愊偟偨unfolded protein傪張棟偟偒傟側偄応崌丄偦傟傜偼ER偐傜僾儘僥傾僜乕儉偵塣偽傟暘夝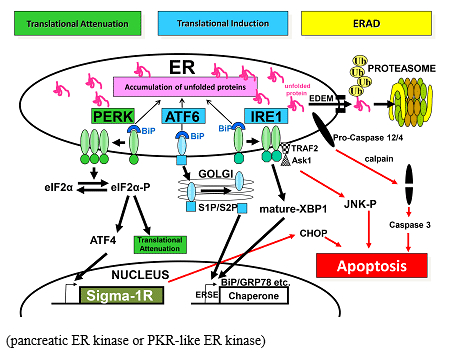 偝傟傞(Bonifacino et al, 1998)丅摐抈敀偺応崌丄僔儍儁儘儞桿摫偵傛傞calnexin傗calreticulin偼unfolded protein偵寢崌偟丄UDP-glucose-glycoprotein glucosyltransferase偲偺娫偵calnexin/calreticulin cycle傪宍惉偡傞(Deprez et al, 2005)丅calnexin/calreticulin cycle偵偁傞摐僞儞僷僋偼丄兛-mannosidase 嘥偵傛傝mannose偑愗傝弌偝傟傞偲丄calnexin/calreticulin傪棧傟丄ER degradation-enhancing兛-mannosidase-like protein乮EDEM乯偲寢崌偟丄unfolded偐斲偐傪尒嬌傔傜傟傞(Molinari et al, 2003)丅EDEM偵傛傝unfolded protein偲擣幆偝傟偨摐抈敀偼translocon傪捠偭偰ER撪偐傜嵶朎幙傊偲塣偽傟(Lee et al, 2004)丄E1-E2-E3 ubiquitin system偵傛傝儐價僉僠儞壔傪庴偗丄26S僾儘僥僆僜乕儉偱暘夝偝傟傞丅
偝傟傞(Bonifacino et al, 1998)丅摐抈敀偺応崌丄僔儍儁儘儞桿摫偵傛傞calnexin傗calreticulin偼unfolded protein偵寢崌偟丄UDP-glucose-glycoprotein glucosyltransferase偲偺娫偵calnexin/calreticulin cycle傪宍惉偡傞(Deprez et al, 2005)丅calnexin/calreticulin cycle偵偁傞摐僞儞僷僋偼丄兛-mannosidase 嘥偵傛傝mannose偑愗傝弌偝傟傞偲丄calnexin/calreticulin傪棧傟丄ER degradation-enhancing兛-mannosidase-like protein乮EDEM乯偲寢崌偟丄unfolded偐斲偐傪尒嬌傔傜傟傞(Molinari et al, 2003)丅EDEM偵傛傝unfolded protein偲擣幆偝傟偨摐抈敀偼translocon傪捠偭偰ER撪偐傜嵶朎幙傊偲塣偽傟(Lee et al, 2004)丄E1-E2-E3 ubiquitin system偵傛傝儐價僉僠儞壔傪庴偗丄26S僾儘僥僆僜乕儉偱暘夝偝傟傞丅
俀丏ER僗僩儗僗僙儞僒乕暘巕 (恾嶲徠)
丂UPR偼ER撪偺unfolded protein偺拁愊傪姶抦偡傞偙偲偐傜巒摦偡傞丅尰嵼傑偱丄ER枌忋偵懚嵼偟丄unfolded protein偺僙儞僒乕偲偟偰PERK丄ATF6丄IRE1偑曬崘偝傟偰偄傞丅
倎丏PERK (pancreatic ER kinase or PKR-like ER kinase)
丂PERK偼ER枌忋偺嘥宆枌娧捠抈敀偱丄N枛抂偺ER撪峯椞堟偼ER僗僩儗僗僙儞僒乕偱偁傝丄C枛抂偼eukaryotic translation initiation factor 2α (eIF2α)傪儕儞巁壔偡傞僙儕儞/僗儗僆僯儞僉僫乕僛妶惈傪帩偮(Shi et al, 1998; Harding et al, 1999)丅偙偺PERK偺僗僩儗僗僙儞僒乕椞堟偼BiP 偑寢崌偟偰偄傞偙偲偱晄妶惈壔偝傟偰偄傞丅ER僗僩儗僗偑惗偠傞偲丄ER撪偵拁愊偟偨unfolded protein乮Accumulation of unfolded proteins乯偼BiP傪PERK偐傜堷偒棧偟丄PERK偺懡検懱壔媦傃帺屓儕儞巁壔傪婲偙偡(Bertolotti et al, 2000)丅儕儞巁壔偝傟偨PERK偼eIF2α偺51埵偺僙儕儞傪儕儞巁壔偡傞丅儕儞巁壔偝傟偨eIF2α(eIF2α-P)偼43S initiation complex偺宍惉傪慾奞偟丄東栿奐巒傪慾奞偡傞 (Translational attenuation)(Shi et al, 1998)丅偙傟偵傛傝丄懡偔偺抈敀偼ER僗僩儗僗壓偱偼嶻惗偑掅壓偡傞偑丄揮幨場巕偺ATF4側偳偼媡偵嶻惗偑忋徃偟丄摿掕偺堚揱巕偺揮幨傪忋徃偝偣傞(Harding, et al, 2000)丅偦傟傜偺侾偮偺GADD34偼protein phosphatase 1 偲暋崌懱(GADD34-PP1)傪宍惉偟丄eIF2α傪嵞傃扙儕儞巁壔偟偰丄抈敀東栿傪尦偵栠偟丄ER僗僩儗僗斀墳偼廔寢偡傞(Novoa et al, 2001)丅
倐丏ATF6
丂ATF6傕ER枌忋偺嘦宆枌娧捠抈敀偱偁傞偑丄偙偺暘巕偵傕旕ER僗僩儗僗壓偱偼BiP 偑撪峯懁偵寢崌偟偰偍傝丄僑儖僕懱堏峴僔僌僫儖傪慾奞偟偰偄傞(Shen et al, 2005)丅ER僗僩儗僗偵嵺偟丄ATF6偼BiP 偲偺寢崌傪夝偒彫朎桝憲偵偰僑儖僕懱偵塣偽傟丄site-1 protease (S1P)媦傃site-2 protease (S2P)偵傛偭偰嵶朎幙懁偺枌娧捠椞堟嬤朤偱愗抐傪庴偗傞丅偱偒偨N枛抂椞堟偼ER枌偐傜棧傟妀偵堏峴偟丄ERSE偵寢崌偡傞偙偲偱BiP傗calreticulin偺桿摫傪懀恑偡傞(Yoshida et al, 1998)丅
們丏IRE1
IRE1傕ER枌忋偺嘥宆枌娧捠偺僙儕儞/僗儗僆僯儞僉僫乕僛偱偁傝丄僄儞僪儕儃僰僋儗傾乕僛(RNase)妶惈傪帩偮丅IRE1偺撪峯椞堟偼PERK偺偦傟偲憡摨惈偑崅偔丄旕僗僩儗僗壓偱偼ER僔儍儁儘儞抈敀偱偁傞BiP偑IRE1偺撪峯懁偵寢崌偟偰偄傞偲峫偊傜傟偰偄傞丅ER撪偵unfolded protein偑弌尰偡傞偲丄BiP偑棧傟丄IRE1偼擇検懱傪宍惉偟RNase妶惈偵傛偭偰帺屓儕儞巁壔傪峴偆丅傎擕椶偺嵶朎偱偼丄偙偺儕儞巁壔偝傟偨IRE1偑XBP1mRNA偺僀儞僩儘儞傪愗傝弌偡偙偲偱僼儗乕儉僔僼僩傪婲偙偝偣丄C枛抂偵揮幨懀恑場巕傪帩偮惉弉偟偨XBP1(mature-XBP1)偑弌尰偡傞(Calfon et al, 2002; Yoshida et al, 2001)丅偙偺惉弉XBP1偼妀偵堏峴偟丄BiP側偳偺僔儍儁儘儞抈敀堚揱巕偺僾儘儌乕僞乕晹埵偵偁傞UPR elements (UPRE)偵寢崌偟丄偙傟傜偺僔儍儁儘儞抈敀桿摫偑峴傢傟傞(Tirasophon et al, 1998; Wang et al, 1998)丅偝傜偵丄惉弉XBP1偼EDEM側偳偺ERAD娭楢場巕偺桿摫傕峴偆(Oda et al, 2006)丅
埲忋偺傛偆偵丄ER僗僩儗僗僙儞僒乕偺妶惈壔偼寢崌偟偰偄偨BiP偑棧傟傞偙偲偵傛傝惗偠傞偲偄偆嫟捠偺儊僇僯僘儉偑憐掕偝傟偰偄傞丅
4乯偆偮昦偺奼嶶僥儞僜儖夋憸傪梡偄偨昦懺専摙
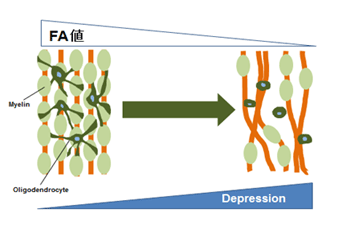 摉尋媶幒偱偼偆偮昦姵幰偍傛傃寬忢幰偺MRI偵傛傞奼嶶僥儞僜儖夋憸傪嶣憸偟丄夝愅傪恑傔偰偄傞丅摿偵丄敀幙偵偍偗傞偆偮昦偵傛傞曄壔偵拲栚偟偰偍傝丄忋婰偺暘巕惗暔妛揑側幚尡偲偺梈崌傪栚巜偟偰偄傞乮恾乯傑偨丄偆偮昦偺惗暔妛揑儅乕僇乕偺妋棫傕栚巜偟偰偄傞丅
摉尋媶幒偱偼偆偮昦姵幰偍傛傃寬忢幰偺MRI偵傛傞奼嶶僥儞僜儖夋憸傪嶣憸偟丄夝愅傪恑傔偰偄傞丅摿偵丄敀幙偵偍偗傞偆偮昦偵傛傞曄壔偵拲栚偟偰偍傝丄忋婰偺暘巕惗暔妛揑側幚尡偲偺梈崌傪栚巜偟偰偄傞乮恾乯傑偨丄偆偮昦偺惗暔妛揑儅乕僇乕偺妋棫傕栚巜偟偰偄傞丅
5乯偆偮昦偵懳偡傞擣抦峴摦椕朄偺幚慔偲壢妛揑専徹
岤惗壢尋乽乽擣抦峴摦椕朄摍偺惛恄椕朄偺壢妛揑僄價僨儞僗偵婎偯偄偨昗弨帯椕偺奐敪偲晛媦偵娭偡傞尋媶乿乮戝栰桾斍挿乯偵暘扴尋媶偲偟偰嶲壛偟丄擣抦峴摦椕朄偺壢妛揑専徹傪忋婰偺奼嶶僥儞僜儖夋憸傪梡偄偨庤朄偱専摙偟偰偄偔丅
俇乯悋柊娭楢幘姵偲擣抦徢偺昦懺夝柧
儗儉悋柊峴摦堎忢徢傪庡偲偡傞悋柊娭楢幘姵偼丄擣抦徢偲偺娭楢偑曬崘偝傟偰偍傝丄偦偺恄宱婎斦偺夝柧偼丄擣抦徢偺憗婜恌抐傗帯椕夘擖偵傕偮側偑傞偙偲偑婜懸偝傟偰偄傞丅偙傟傑偱丄摉奩椞堟偱偼恄宱惗棟妛揑庤朄傪拞怱偵尋媶偑側偝傟偰偒偰偄傞偑丄崱屻偼暘巕惗暔妛揑庤朄丄擼宍懺丒婡擻夋憸丄恄宱惗棟妛揑庤朄傪桳婡揑偵寢傃偮偗丄昦懺夝柧偵傾僾儘乕僠偡傞偙偲傪栚巜偟偰偄傞丅
俈乯帪寁堚揱巕傪梡偄偨晄柊丄偣傫栂儌僨儖偺昦懺夝柧
晄柊丄偣傫栂偺敪徢梫場偲偟偰丄悋柊妎惲儕僘儉偺攋抅偑堦偮偺戝偒側梫場偲峫偊傜傟偰偄傞丅惗暔偺儕僘儉偼懱撪帪寁偵傛傝婯掕偝傟丄僸僩傕椺奜側偔懱撪帪寁偑惓忢偵摥偔偙偲偵傛傝抧媴偺俀係帪娫儕僘儉偵揔墳偡傞偙偲偑偱偒傞丅帪寁堚揱巕偼偙偺懱撪帪寁偺婡峔傪幚嵺偵摦偐偟偰偍傝丄偦偺婡擻傪昡壙偡傞偙偲偵傛傝丄晄柊丄偣傫栂偺幘昦儌僨儖偵墳梡偱偒傞偲峫偊傜傟傞丅暘巕惗暔妛揑傾僾儘乕僠傕梡偄丄偙偺婡峔傪柧傜偐偵偡傞偙偲傪恑傔偰偄偔丅